ASHKA kinase

Murein recycling is a process in which microorganisms recover peptidoglycan degradation products in order to utilize them in cell wall biosynthesis or basic metabolic pathways. Methanogens like Methanopyrus kandleri contain pseudomurein that differs from bacterial murein in composition and branching. We have solved the crystal structure of the ASKHA superfamily sugar kinase MK0840 from Methanopyrus kandleri [2], that shows high similarity to bacterial anhydro-N-acetylmuramic acid kinase involved in murein recycling. We could observe local conformational changes in the nucleotide-binding site between the apo and holo form upon nucleotide binding. Further insight was gained into domain movements and putative active-site residues were identified.
Related PDB: 4BG8, 4BG9, 4BGA, 4BGB
- M. Schacherl, S. Waltersperger, U. Baumann, Structural characterization of the ribonuclease H-like type ASKHA superfamily kinase MK0840 from Methanopyrus kandleri, Acta Crystallogr D Biol Crystallogr 69 (2013) 2440-2450
Archaemetzincins
Archaemetzincins are metalloproteases occurring in archaea and some mammalia. They are distinct from all the other metzincins by their extended active site consensus sequence HEXXHXXGXXHCX(4)CXMX(17)CXXC featuring four conserved cysteine residues. Very little is known about their biological importance and structure-function relationships. We could solve crystal structures of the archaemetzincin AfAmzA (Uniprot O29917) from Archaeoglobus fulgidus [1] and Methanopyrus kandleri [2], revealing a metzincin architecture featuring a zinc finger-like structural element involving the conserved cysteines of the consensus motif. The active sites in all structures are occluded to different extents rendering the enzymes proteolytically inactive against a large variety of tested substrates. Owing to the different ligand binding there are significant differences in active site architecture, revealing a large flexibility of the loops covering the active site cleft. The crystal structures of AmzA provide the structural basis for the lack of activity in standard proteolytic assays and imply a triggered activity onset upon opening of the active site cleft.
Related PDB: 3ZVS, 4A3W, 4AXQ, 2X7M
- C. Graef, M. Schacherl, S. Waltersperger, U. Baumann, Crystal structures of archaemetzincin reveal a moldable substrate-binding site, PLoS One 7 (2012) e43863.
- S. Waltersperger, C. Widmer, M. Wang, U. Baumann, Crystal structure of archaemetzincin AmzA from Methanopyrus kandleri at 1.5 A resolution, Proteins 78 (2010) 2720-2723.
ATP-dependent metalloprotease FtsH
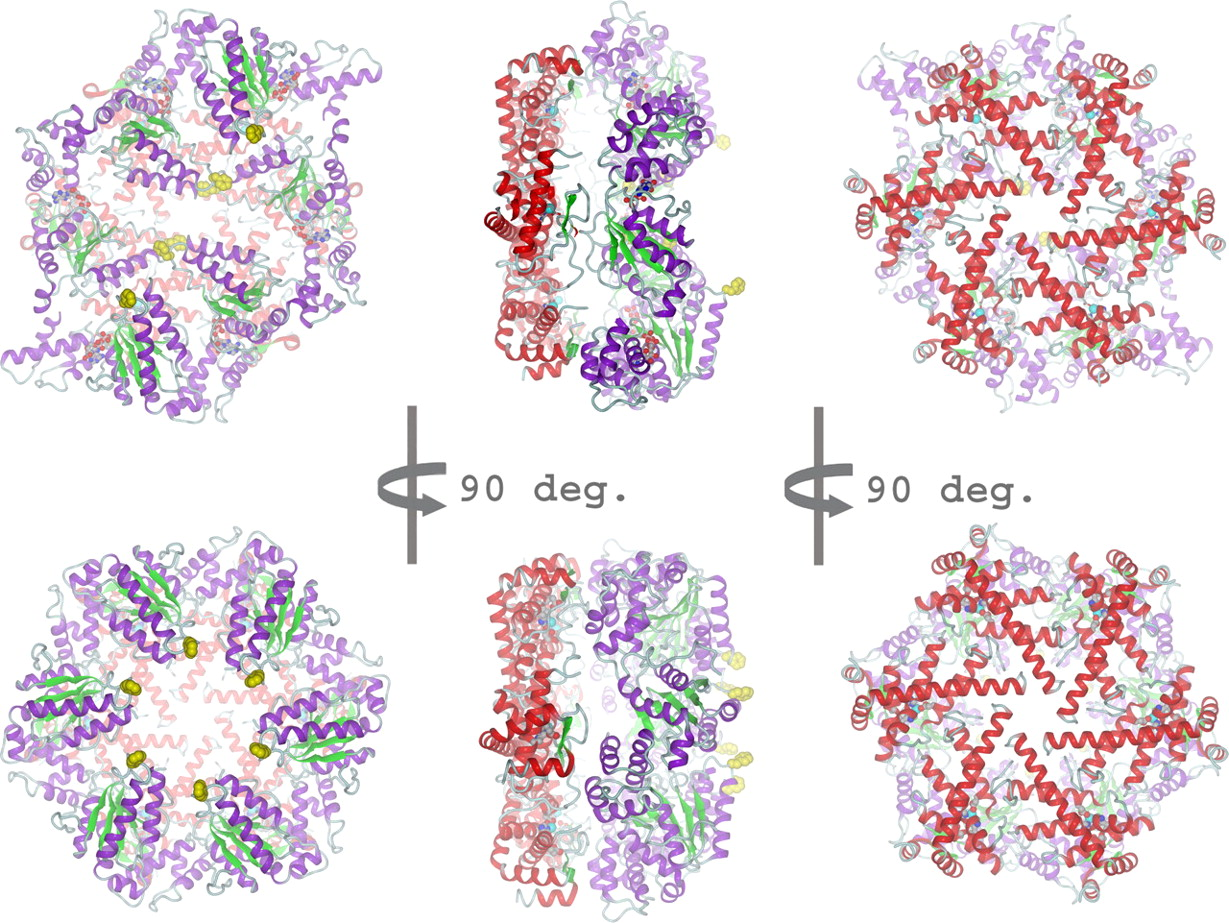
The ATP-dependent integral membrane protease FtsH is universally conserved in bacteria. Orthologs exist in chloroplasts and mitochondria, where in humans the loss of a close FtsH-homolog causes a form of spastic paraplegia. FtsH plays a crucial role in quality control by degrading unneeded or damaged membrane proteins, but it also targets soluble signaling factors like sigma(32) and lambda-CII. We could solve the crystal structure of a soluble FtsH construct that is functional in caseinolytic and ATPase assays [1,2]. The molecular architecture of this hexameric molecule consists of two rings where the protease domains possess an all-helical fold and form a flat hexagon that is covered by a toroid built by the AAA domains. The active site of the protease classifies FtsH as an Asp-zincin, contrary to a previous report. The different symmetries of protease and AAA rings suggest a possible translocation mechanism of the target polypeptide chain into the interior of the molecule where the proteolytic sites are located.
- C. Bieniossek, T. Schalch, M. Bumann, M. Meister, R. Meier, U. Baumann, The molecular architecture of the metalloprotease FtsH, Proc Natl Acad Sci U S A 103 (2006) 3066-3071.
- C. Bieniossek, B. Niederhauser, U. M. Baumann, The crystal structure of apo-FtsH reveals domain movements necessary for substrate unfolding and translocation, Proc Natl Acad Sci U S A 106 (2009) 21579-21584.
PPEP 1

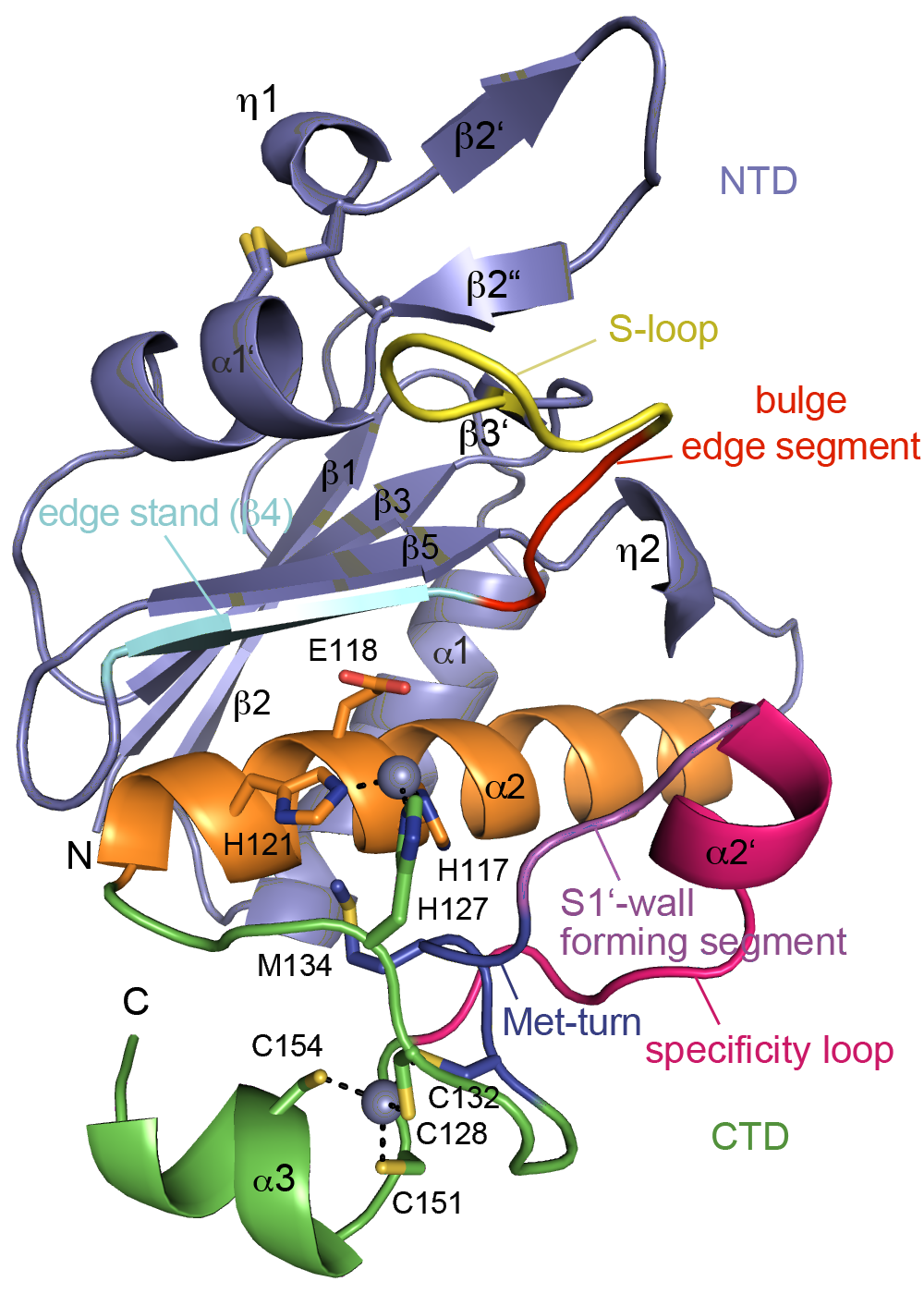
Clostridium difficile is a pathogenic bacterium causing gastrointestinal diseases from mild diarrhea to toxic megacolon. In common with other pathogenic bacteria, C. difficile secretes proteins involved in adhesion, colonization, and dissemination. PPEP-1 (also called Zmp1) is an extracellular metalloprotease showing a unique specificity for Pro-Pro peptide bonds. The endogenous substrates of PPEP-1 are two surface proteins implicated in adhesion of C. difficile to surface proteins of human cells. Thus, PPEP-1 is believed to be involved in the regulation of the adhesion-motility balance of C. difficile. We could solve crystal structures of PPEP-1 from C. difficile in its unbound and peptide-bound forms [1]. The structure analysis revealed a fold similar to Bacillus anthracis lethal factor. Crystal structures in the open and closed conformation of the S-loop shed light on the mode of binding of the substrate, and reveal important residues for substrate recognition and the strict specificity of PPEP-1 for Pro-Pro peptide bonds.
Related PDB: 5A0P, 5A0S, 5A0R, 5A0X
1. M. Schacherl, C. Pichlo, I. Neundorf, U. Baumann, Structural basis of proline-proline peptide bond specificity of the metalloprotease Zmp1 implicated in motility of Clostridium difficile, Structure 23(9) (2015) 1632–1642